Диагностика и лечение первичных Т-клеточных лимфом кожи
Первичные Т-клеточные кожные лимфомы представляют собой гетерогенную группу лимфопролиферативных заболеваний, характеризующуюся клональной пролиферацией Т-лимфоцитов в коже. Они составляют 75–80 % всех кожных лимфом. Разграничение первичных лимфом кожи и вторичных ее поражений при других лимфомах требует тщательного анализа клинической картины, патологических, иммунологических и молекулярно-генетических данных. Первичные лимфомы кожи отличаются от нодальных лимфом характером течения, прогнозом и подходами терапии. Это обстоятельство вызвало необходимость создания подробной классификации, отражающей весь спектр первичных кожных лимфом. В 2005 г. на основе классификации ВОЗ для опухолей гемопоэтической и лимфоидной тканей [1] и классификации лимфом кожи Европейской организации по исследованию и лечению рака (EORTC) [2] создана ВОЗ/EORTC-классификация кожных лимфом, наиболее полно охватывающая весь спектр этих заболеваний [3].
Т- и NK-клеточные лимфомы кожи
- Грибовидный микоз (ГМ):
— фолликулотропный ГМ;
— педжетоидный ретикулез;
— синдром гранулематозной вялой кожи.
- Синдром Сезари.
- Первичные кожные CD30+-лимфопролиферативные заболевания:
— первичная анапластическая крупноклеточная лимфома кожи;
— лимфоматоидный папулез.
- Подкожная панникулитоподобная Т-клеточная лимфома.
- Экстранодальная NK/Т-клеточная лимфома, назальный тип.
- Первичная кожная периферическая Т-клеточная лимфома, неуточненная.
- Первичная кожная агрессивная эпидермотропная CD8+ Т-клеточная лимфома.
- Кожная γ/δ-Т-клеточная лимфома.
- Первичная кожная CD4+ плеоморфная Т-клеточная лимфома из клеток малых и средних размеров.
Самыми распространенными подтипами являются: ГМ, синдром Сезари, первичная анапластическая крупноклеточная лимфома кожи и лимфоматоидный папулез. Они составляют приблизительно 95 % всех Т-клеточных кожных лимфом.
ГМ — первичная эпидермотропная Т-клеточная лимфома кожи, отличительной чертой которой является пролиферация Т-лимфоцитов малых и средних размеров с церебриформными ядрами. Термин «грибовидный микоз» в настоящее время принято использовать только для классического варианта микоза Алибера–Базена, характеризующегося поэтапной эволюцией пятен, бляшек и узлов, или для вариантов со схожим клиническим течением.
Это наиболее часто встречающаяся Т-клеточная опухоль кожи, составляющая 1 % всех неходжкинских лимфом и 50% Т-клеточных кожных лимфом. Средний возраст заболевших — 57 лет, соотношение мужчин и женщин — 2 : 1 [4]. ГМ, особенно на ранних стадиях, может протекать «под маской» различных доброкачественных кожных процессов, таких как хроническая экзема, аллергический контактный дерматит или псориаз. Для начальных кожных проявлений характерна локализация на ягодицах и других защищенных от солнца областях. Заболевание обычно течет благоприятно, медленно прогрессируя в течение нескольких лет или даже десятилетий. Кожные морфологические элементы постепенно эволюционируют от пятен или бляшек до опухолевых узлов с признаками изъязвления. На поздних стадиях заболевания в патологический процесс могут вовлекаться лимфатические узлы и внутренние органы.
Гистологическая картина на ранних стадиях ГМ неспецифична и может быть схожа с таковой при доброкачественном воспалительном дерматозе: отмечаются периваскулярные инфильтраты в сочетании с псориазоформной гиперплазией эпидермиса [5]. Для бляшечной стадии характерен плотный полосовидный инфильтрат в верхней части дермы, содержащий высокий процент церебриформных лимфоцитов с выраженным эпидермотропизмом. Внутриэпидермальные скопления атипичных лимфоцитов (микроабсцессы Потрие) являются характерной чертой этой стадии, но встречаются лишь в 10 % случаев [6]. С прогрессированием в опухолевую стадию эпидермотропизм исчезает, а инфильтрат, состоящий из церебриформных лимфоцитов малых, средних и крупных размеров, становится диффузным и может проникать в подкожную жировую клетчатку.
Опухолевые клетки при ГМ имеют фенотип зрелых Т-лимфоцитов памяти (CD3+, CD4+, CD45RO+, CD8-). Редко может наблюдаться фенотип CD4-, CD8+. Клиническое течение и прогноз в таких случаях не отличаются от классического варианта, и, следовательно, их не следует рассматривать отдельно. О наличии аберрантного фенотипа при ГМ говорит утрата пан-Т-клеточных антигенов CD2, CD3, CD5, CD7, что во многих случаях является важным дополнением к диагнозу [7].
Прогноз при ГМ напрямую зависит от стадии заболевания, характера и распространенности кожного процесса, а также наличия внекожных поражений.

|
| Таблица 1. TNMB классификация грибовидного микоза |
В 1978 г. Национальным институтом рака США была предложена TNMB (tumor, node, metastasis, blood) — классификация кожных Т-клеточных лимфом, которая применима для определения стадий ГМ (табл. 1, 2) [8]. Данные о пятилетней выживаемости при грибовидном микозе/синдроме Сезари (ГМ/СС) в зависимости от стадии заболевания следующие: IA — 96 %, IB / IIA — 73 %, IIB / III — 44 %, IV — 27 %.
Кроме классической формы ГМ, имеется несколько вариантов этого заболевания с необычными клиническими и/или гистологическими характеристиками. Из них в классификации ВОЗ/EORTC выделены три клинико-патологических варианта: фолликулотропный ГМ, педжетоидный ретикулез и синдром гранулематозной «вялой» кожи [3].
|
| Таблица 2. Стадии грибовидного микоза по классификации TNMB (США, 1978) |
Фолликулотропный ГМ характеризуется наличием фолликулотропного и часто неэпидермотропного инфильтрата. Клинически заболевание может проявляться фолликулярными папулами, бляшками и иногда опухолями, локализующимися чаще всего на голове и шее и сопровождающимися алопецией. Гистологически выявляется плотный периаднексальный (перифолликулярный) инфильтрат, состоящий из малых и средних лимфоидных клеток с церебриформными ядрами. Эпидермотропизм может отсутствовать. Волосяные фолликулы часто кистозно расширены, возможна муцинозная дегенерация фолликулярного эпителия.
Педжетоидный ретикулез (моноочаговая форма Ворингера–Колоппа) является благоприятно протекающей формой ГМ и характеризуется наличием одного очага в виде псориазоформной бляшки, локализующейся на нижних конечностях. Гистологическая картина характеризуется акантотическим эпидермисом, содержащим «спонгиоформный» инфильтрат из средних и крупных лимфоидных клеток с вакуолизированной цитоплазмой, расположенных поодиночке или скоплениями.
К одним из самых редко встречающихся вариантов ГМ относится синдром гранулематозной «вялой» кожи, характеризующийся клональной пролиферацией Т-лимфоцитов и дегенерацией эластических волокон в дерме. Заболевание клинически проявляется развитием в крупных складках своеобразных изменений кожи в виде появления складчатых, инфильтрированных и лишенных эластичности образований. Гистологическая картина характеризуется наличием плотного диффузного инфильтрата, состоящего из малых и средних лимфоидных клеток без выраженной атипии ядер, и отсутствием эластических волокон в сосочковой и сетчатой частях дермы. Специфическим гистологическим признаком данного заболевания считается наличие в инфильтрате многоядерных гигантских клеток, имеющих от 20 до 30 концентрически расположенных ядер. В их цитоплазме могут находиться лимфоциты, а также остатки эластических волокон.
Синдром Сезари характеризуется триадой признаков: эритродермия, лимфаденопатия и наличие опухолевых Т-лимфоцитов (клеток Сезари) в коже и периферической крови [9].
Клинически синдром Сезари характеризуется развитием эритродермии с генерализованным зудом кожного покрова, увеличением периферических лимфатических узлов, наличием ладонно-подошвенного гиперкератоза и ониходистрофии. Интернациональным обществом по кожным лимфомам были предложены следующие критерии для диагностики синдрома Сезари: абсолютное количество клеток Сезари в периферической крови не меньше 1000 в мм3, увеличение соотношения CD4/CD8 лимфоцитов более чем в 10 раз и потеря Т-клеточных антигенов (CD2, CD3, CD4, CD5), подтверждение Т-клеточной клональности в периферической крови молекулярными или цитогенетическими исследованиями [10].
Гистологические изменения при синдроме Сезари сходны с таковыми при ГМ, однако эпидермотропизм может быть менее выражен.
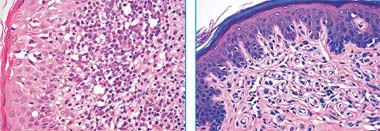
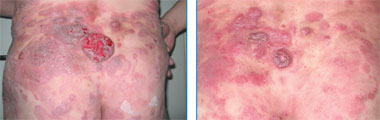
Терапия ГМ/СС также зависит от стадии заболевания. На ранних стадиях, при локальных изменениях кожи, эффективна РUVA-терапия, которая позволяет получить полную ремиссию в подавляющем большинстве случаев [11–13]. При I и II стадиях PUVA-терапия может быть использована в комбинации с интерфероном α [14–16]. Применение агрессивных методов лечения (химио- и лучевая терапия) на ранних стадиях не меняет прогноз и, следовательно, неоправдано [17]. При распространенном поражении локальная лучевая терапия, тотальное облучение кожи (ТОК), экстракорпоральный фотоферез позволяют контролировать течение болезни, но имеют ограниченную доступность [18–21]. Когда заболевание становится резистентным к указанным методам лечения, используется комбинированная химиотерапия, однако вне зависимости от ее варианта продолжительность эффекта обычно не превышает 1 года [22–26]. В последнее время в лечении ГМ/СС все большее распространение получает применение биологических препаратов, механизм действия которых основан на специфическом связывании с различными антигенами на мембране опухолевых клеток. К ним относятся интегрированный протеин — Ontak и анти-CD52-моноклональное антитело — алемтузумаб (Кэмпас). Ontak является конъюгатом токсина дифтерии с интерлейкином-2, который после связывания с рецептором к интерлейкину-2 (СD25) подвергается эндоцитозу с последующим высвобождением внутри клетки дифтерийного токсина. Результатом этого процесса является нарушение синтеза белка и в конечном итоге апоптоз Т-лимфоцитов [27]. Кэмпас представляет собой гуманизированное IgG1-моноклональное антитело, специфически связывающееся с CD52-антигеном. Эффекторный механизм Кэмпаса изучен не до конца, он, вероятно, основан на антителозависимой клеточной цитотоксичности [28, 29], комплемент-обусловленном клеточном лизисе [30, 31] и апоптозе [32]. Опухолевые Т-лимфоциты экспрессируют на своей поверхности большое количество молекул CD52 (около 500 000 молекул на лимфоцит), и интенсивность экспрессии CD52 напрямую коррелирует с клиническим эффектом [33, 34]. Основанием для использования Кэмпаса при ГМ является его успешное применение при других Т-клеточных опухолях, например, при Т-клеточном пролимфоцитарном лейкозе [35]. По данным одного из наиболее крупных исследований по использованию Кэмпаса в терапии ГМ/СС, в которое было включено 22 больных со II–IV стадиями заболевания, ранее получавших другие виды лечения, общий ответ на терапию составил 55 %, полная ремиссия достигнута в 32 % случаев. Если предшествующее лечение включало в себя не более двух режимов терапии, общий ответ составлял 80 %. Медиана выживаемости без прогрессии для 12 больных, ответивших на лечение, составила 12 мес [36]. Клиническая картина до и после терапии ГМ препаратом Кэмпас представлена на рис. 1–4.
|
|
Рис. 1. Разрушение зоны эпидермально-дермального стыка. Распространение инфильтрата в эпидермис с образованием абсцессов Потрие. Рис. 2. Единичные лимфоидные клетки среди эпителиоцитов базального слоя эпидермиса (после терапии алемтузумабом) |
Первичные CD30+-лимфопропролиферативные заболевания кожи занимают второе место по частоте возникновения среди всех Т-клеточных лимфом кожи. Они включают анапластическую крупноклеточную лимфому кожи и лимфоматоидный папулез. Их объединяющим признаком является экспрессия опухолевыми клетками CD30 — рецептора, принадлежащего к группе рецепторов фактора некроза опухолей.
Первичная анапластическая лимфома кожи чаще развивается у лиц мужского пола, в основном в возрасте старше 60 лет. Клинически определяются обычно один или несколько узлов (в том числе подкожных), имеющих тенденцию к изъязвлению. Наиболее частая локализация высыпаний — верхние и нижние конечности. Гистологическая картина представлена диффузными инфильтратами в дерме и подкожно-жировой клетчатке, состоящими из клеток с анапластической или иммунобластной морфологией. Эпидермотропизм не характерен. Опухолевые клетки экспрессируют CD30, а также один или несколько пан-Т-клеточных антигенов (CD2, CD3, CD4, CD5). Экспрессия антигена эпителиальных мембран (EMA) и ALK-протеина обнаруживается крайне редко, что отличает первичную кожную анапластическую лимфому от нодальной анапластической лимфомы с поражением кожи [37].
Первичная анапластическая лимфома кожи имеет, как правило, доброкачественное течение. Пятилетняя выживаемость составляет около 90 %. Лучевая терапия или хирургическое удаление применяются в случае единичных очагов. У больных с множественными высыпаниями проводится лечение малыми дозами метотрексата (5–20 мг в неделю) или лучевая терапия. Только быстропрогрессирующее течение заболевания с внекожным распространением требует назначения полихимиотерапии.
|
|
Рис. 3. Множественные пятна, бляшки и опухолевые образования с элементами изъязвления на коже (до начала лечения). Рис. 4. Вид после терапии алемтузумабом |
Лимфоматоидный папулез — хроническое лимфопролиферативное заболевание кожи, характеризующееся рецидивирующим течением с повторными высыпаниями самопроизвольно разрешающихся папулезных элементов с гистологическими признаками CD30+-лимфомы. Выделяют три гистологических варианта заболевания: тип А, тип В и тип С.
Тип А характеризуется кожным инфильтратом состоящим из крупных атипичных клеток, напоминающих клетки Рида–Штернберга.
Тип В гистологически напоминает картину ГМ. Анапластические клетки встречаются в малом количестве или отсутствуют.
Тип С характеризуется мономорфным инфильтратом или наличием крупных кластеров CD30+ Т-лимфоцитов. Наиболее эффективным методом лечения является назначение малых доз метотрексата, особенно в случае распространенных высыпаний и частых обострений [3].
Таким образом, диагностика и лечение первичных Т-клеточных кожных лимфом требуют учета многих факторов и использования широкого спектра современных методов обследования (гистологических, иммунологических, молекулярно-генетических), позволяющих правильно определить прогноз и оптимизировать терапию.
По вопросам литературы обращайтесь в редакцию.
В. А. Доронин
ЦКБ № 2 им. Н. А. Семашко, Москва